The FDA’s Expedited Review Process: The Need for Speed
February 2015

FDA Drug Development Timeline
It takes an average of 12 years for a drug to progress from early laboratory discovery to approval for human use (Figure 1). The FDA works to protect public health by balancing the requirements for extensive safety and efficacy data prior to approval, and the need to expeditiously issue approval decisions to ensure medicines that could save or dramatically improve patients’ lives are available as soon as possible.
The FDA currently utilizes 4 programs to expedite reviews of drugs for serious and life threatening diseases. Each program has specific requirements and benefits, and can be used in various combinations. At the start of any drug development program, an in-depth understanding of these options is critical to ensuring the most efficient path to approval. In the Fall 2014 Tribune, the newest FDA program was discussed in the article,
“The FDA’s ‘Breakthrough’ Designation: Two Years of Fun”. This article examines
the other FDA expedited review processes: Accelerated Approval, Priority Review,
and Fast Track in relation to the Breakthrough Designation.
Figure 1: Drug Discovery and Development Timeline
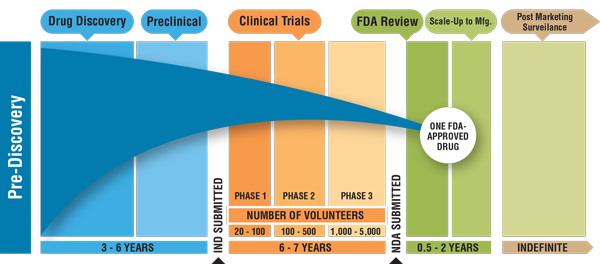
Credit: cancerprogressreport.org [Internet]. Philadelphia: American Association for Cancer Research; ©2011.
Available from http://www.cancerprogressreport-digital.org/cancerprogressreport/2011?pg=1#pg1.
Accelerated Approval Allows the Use of Surrogate Endpoints
AIDS Necessitated the Need for Expedited Review Legislation
In response to the AIDS crisis in the late 1980’s, the FDA drafted initiatives to accelerate drug delivery and cut costs for new drugs treating serious and life-threatening illnesses and conditions, granting approval to drugs after extended phase 2 trials. In 1992, in response to a push by AIDS advocates to make the investigational anti-AIDS drug azidothymidine (AZT) accessible, the FDA enacted “Subpart H” commonly referred to as Accelerated Approval; giving rise to expedited review of drugs by the FDA. This legislation allowed new drug applications (NDAs) to be approved based on surrogate endpoints in clinical trials including 1) markers that would be expected to confer a clinical benefit such as improved overall survival, prolonged suppression of HIV viral load in HIV, or tumor shrinkage in cancer, or 2) an intermediate clinical endpoint; a measurement of a therapeutic effect considered reasonably likely to predict clinical benefit, such as an effect on irreversible morbidity or mortality (IMM). Unlike the other expedited review processes discussed in this article, there is no formal application process for designating a product for development through the Accelerated Approval pathway. Drug sponsors that decide to pursue this pathway meet with the FDA early in drug development and agree on the following criteria:
- The unmet need that exists in the patient population being studied
- The surrogate endpoints that will be assessed
- The magnitude of benefit that must be observed using the agreed-upon surrogate endpoint
- Making decisions and taking action based on factual analysis, balanced with experience and intuition
In controlled randomized trials, the FDA and the sponsor could also agree than an interim analysis of data using a surrogate endpoint such as progression-free survival (PFS) is required. If sufficient efficacy is observed during interim analysis, Accelerated Approval could be granted or the FDA could decide that a trial should be completed using a traditional clinical endpoint, for conversion to full approval. For Accelerated Approval using surrogate endpoints, drug sponsors are required to perform Phase 4 confirmatory trials and post-marketing studies to verify the predicted clinical benefit. If clinical benefit is shown, the drug receives traditional full approval; however, if no clinical benefit is shown, the drug is subject to expedited withdrawal and required to be immediately removed from the market.
Acceleration of Oncology Drug Approval
Accelerated Approval has been vital in expediting access primarily to oncology drugs for cancer patients. From 2000 to 2012, 53% of Accelerated Approvals were for cancer drugs, 18% were for HIV and 29% for “Other”, with cardiovascular leading the “Other” category. As of July 1, 2010, 35 oncology products had obtained Accelerated Approval for 47 indications, and 26 were converted to full approval with an average time of conversion of 4.7 years. Following post-marketing analysis, 3 oncology drugs that were granted accelerated approval have been withdrawn or relabeled because of unexpected safety or apparent lack of efficacy, however, the majority of Accelerated Approvals have confirmed clinical benefit and have been revered as providing life-saving or life-prolonging medicines available to seriously ill patients.
Priority Review Shortens FDA Review Timelines
In 1992, under Prescription Drug User Fee Act (PDUFA), the FDA agreed to improve drug review timelines, and created a two-tiered system of review times, Priority Review and Standard Review. To improve drug review timelines, the FDA grants a Priority Review designation to applicants (for new or supplemental applications), for drugs that 1) treat a serious condition and 2) would provide a significant improvement in the safety or efficacy for a serious condition. Priority Review shortens the target period for FDA review from 10 months to 6 months, and this designation is often used in combination with other expedited review processes.
Fast Track Designation for Drugs with Potential to Address Unmet Medical Needs
Legislation and Requirements for Fast Track Designation
The expedited process “Fast Track” was implemented under the FDA Modernization Act (FDAMA) of 1997, and Fast Track Designation targets drugs that are intended 1) to treat a serious condition and 2) for which data demonstrate the potential to address an unmet medical need. Fast Track addresses the need to approve treatment for a broad range of serious diseases including AIDS, Alzheimer’s, cancer, epilepsy, and diabetes. An application for the Fast Track designation can be submitted at any time during the drug development process and can use preclinical or clinical data to show potential to address an unmet medical need, and the FDA must respond to the application within 60 days. With Fast Track designation, drug sponsors benefit from the opportunity for early and frequent interactions with the FDA review team and from the “rolling review” process where portions of an application can be submitted for review prior to submitting the complete application. Most applicants that are granted the Fast Track designation are also eligible for Priority Review.
The FDA Safety and Innovation Act (FDASIA) of 2012 amended the Fast Track designation to include the Generating Antibiotics Incentives Now Act (GAIN Act), which provides incentives to help bring new antibiotics and other antimicrobials to market. A drug with particular promise can be designated as a Qualified Infectious Disease Product (QIDP) by authority of the GAIN Act, and is then eligible for Fast Track designation and Priority Review. In 2014, the Center for Drug Evaluation and Research (CDER) at the FDA approved 4 new antibiotics with this designation, the first 4 QIDP-designated novel new drugs approved.
Fast Track of Ebola Vaccines
Recently the FDA has been under pressure to Fast Track experimental drugs and vaccines for the Ebola virus, of which there is no known cure, following the spread of the virus in Western Africa with over 8,795 reported deaths as of January 27, 2015, according to the Center for Disease Control and Prevention (CDC). On October 6, 2014 the FDA approved the use of an experimental antiviral drug which has successfully treated Ebola in lab tests. The drug has also been tested by the CDC and the National Institutes of Health (NIH), though it is not expected to win approval for wide public use until late 2016. Another drug produced by a Canadian drug-maker, has also been approved for the Fast Track designation and has been used to treat a patient in Atlanta.
Breakthrough Therapy for Drugs with Substantial Superiority
FDASIA also introduced the Breakthrough Therapy designation into the FDA portfolio of expedited programs to address new trends in drug discovery and development, particularly targeted therapies (including biomarkers), often paired with companion diagnostics, for treatment of cancer, genetic diseases, and increasingly other diseases. The Breakthrough Therapy designation is similar to Fast Track and Accelerated Approval in that it requires the investigational drug be used for a serious or life-threatening disease or condition. Breakthrough Therapy and Fast Track also both allow “rolling review” of drug development material being submitted to the FDA. Breakthrough Therapy designation differs from the other expedited review processes by requiring the use of a clinically significant endpoint that demonstrates substantial superiority of the drug over available therapies. Fast Track allows the use of clinical or nonclinical data demonstrating the potential to address an unmet medical need and Accelerated Approval allows the use of surrogate endpoints that provide reasonable advantage over available therapies. The Breakthrough Therapy Designation offers more expansive benefits than the other expedited processes in that once a drug receives the Breakthrough Therapy designation it is assigned an FDA committee which meets regularly with the sponsor to devise the most efficient way to generate additional safety and efficacy data to move development forward. It is an “all hands on deck” approach, with frequent communication between the drug developer and the FDA, at the division level and across all levels of FDA management.
Breakthrough Designation – a Popular Choice
With these benefits established, Breakthrough Therapy designation rapidly became a popular choice in drug development pathways and Dr. Janet Woodcock, Director of CDER, has stated that the volume of requests for Breakthrough Therapy designation came as a surprise. Between July 2012 and December 2013, the FDA received 135 requests for this designation, 41% of which were for cancer therapies. According to Dr. Woodcock, failure of the preliminary clinical data to suggest a substantial benefit over existing therapy has been a major reason for denial. In situations where some standard-of-care therapy exists, a distinction between small, incremental improvements, and game-changing effects must be made in order for Breakthrough Therapy designation to be granted.
Figure 2: Overview of the Food and Drug Administration's (FDA’s) Expedited Review Drug Approval Programs
| |
Fast track |
Accelerated approval |
Priority review |
Breakthrough therapy |
| Date established |
1988 |
1992 |
1992 |
2012 |
| Qualifying criteria |
- Must be intended to treat a serious condition or a drug designed as a Qualified Infectious Disease Product (QIDP)
- May address an unmet medical need
- Supporting data can be clinical or nonclinical
|
- Must treat a serious condition
- Early evidence shows substantial improvement over existing therapies
- May use surrogate endpoints to demonstrate clinical benefit
|
- Must treat a serious condition or a drug designated as a QIDP or receive a priority review voucher.
- Provides significant improvement in safery or effectiveness over existing therapies
|
- Must treat a serious condition
- Early evidence shows substancial improvement over existing therapies
- Supporting data must be clinical
|
| Time frame for application and FDA response |
Can be requested with an investigational new drug (IND) submissioni or any point after applying. The FDA has sixty days to respond to the request. |
No formal process. Drug sponsors discuss the possibility with the FDA during drug development and decide the planned endpoint. |
Requested at time of drug approval application. The FDA has sixty days to respond to request. |
Can be requested with IND submission or any point after applying. The FDA has sixty days to respond to request. |
| Key program features |
- Earier and more frequent communication with the FDA during development
- Rolling review of application
- Designation may be withdrawn if drug no longer meets qualifying criteria
|
- Approval is granted on a conditional basis. Drug sponsor must conduct post approval confirmatory trials to confirm clinical benefits
- Drug is subject to expedited withdrawal
|
- Drug review process is shortened to six months (from the standard ten months)
|
- All fast-track designation features
- Intensive FDA guidance throughout development process, involving senior FDA officials
- Designation may be withdrawan if drug no longer meets qualifying criteria
|
Source information in this table was adapted from the FDA's "Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics" (May 2014)
Break-down of FDA Expedited Review Designations Granted in 2014
Each of the 4 expedited approval programs has helped improve the speed of the development and/or approval process of numerous drugs and brought important, often life-saving medications to the market, and ultimately patients, as quickly as possible. According to CDER’s “Novel New Drugs 2014 Summary:”
- 66% of the novel new drugs (27 of 41 or 66%) approved by the FDA in 2014 were designated in one or more of the expedited review programs
- 20% used Accelerated Approval
- 61% used Priority Review
- 41% used Fast Track
- 22% had a Breakthrough Therapy designation.
Included in the 41 drugs approved were 8 new drugs for treating patients with various types of cancer, 4 new drugs to treat type-2 diabetes, 4 new antibiotics to treat serious infections, and 2 new products to treat patients with hepatitis C. The future of the expedited review process is likely to continue to evolve as new diseases arise and new technology (ie., targeted therapies influenced by genomics and biomarkers) becomes available.
About the Author
Dr. Shahza Somerville is a Medical Writer and Clinical Research Specialist at TRI. She holds a Ph.D. in neuroscience and has previously done extensive research on the neurotoxicology of antipsychotic drugs at the Maryland Psychiatric Research Center and on countermeasures for chemical defense at Walter Reed Army Institute of Research (WRAIR).
Dr. Jessica Holden Kloda is Associate Director of Regulatory Affairs at TRI. She has over 15 years of experience in biomedical and clinical research. She holds a Ph.D. in Molecular and Cellular Pharmacology from the University of Wisconsin at Madison and the Regulatory Affairs Certification (RAC).


